THE NEW ANNEX 15: A COMPREHENSIVE ANALYSIS

The draft of the revision of Annex 15 was published in February 2014. Just recently it was published as final document and came into operation on 1st October 2015.
What are the changes?
A surprise was that the revised Annex 15 may also be used as supplementary optional guidance for active substances, (but) without introduction of additional requirements of Part II EU GMP Guide. In general, there is a greater distinction between the terms qualification and validation. Utilities now are part of the qualification activities.
A general chapter as well as subsections to the chapters "Qualification" and "Process Validation" were added. The topics verification of transportation, validation of packaging, qualification of utilities and validation of test methods are also new. The chapter "Revalidation" was replaced by the chapter "Re-qualification".
Principle
A life cycle of the product and process that should accompany qualification and validation is already mentioned in the part "Principle". As a sort of extension it is now pointed out that changes should also assess the influence on the control strategy. Furthermore, it is indicated that computerised systems should be validated according to the requirements of Annex 11. The relevant concepts presented in ICH Q8-Q11 should also be taken into account.
A completely new, general part ("General") was added. It states that decisions on the scope and extent of validation and qualification should be based on a justified and documented risk assessment as part of a quality risk management approach. The principles in ICH Q8-11 formerly mentioned are dropped as compared to the draft. Now they are only mentioned in the part "Principle". The last part of this chapter states that data obtained from sources outside of the manufacturers own programmes may be used provided that this approach has been justified and that there is adequate assurance that controls were in place throughout the acquisition of such data. New, as compared to the draft, is the explicit statement that retrospective validation is no longer considered an acceptable approach.

Recommendation
6/7 October 2026
DoE - Live Online Training
Organising and Planning for Qualification and Validation
The first chapter originally named "Planning for Validation" now is called "Organising and Planning for Qualification and Validation". It is stressed that the life cycle of facilities, equipment, utilities, process and product should be taken into consideration and that the personnel should be trained suitably and should follow approved procedures. Validation personnel should report as defined in the internal quality system although this may not necessarily be to a quality management or a quality assurance function. However, there should be appropriate quality oversight over the whole validation life cycle.
Next, the validation master plan (VMP) is addressed. The requirements haven't changed much. (Unfortunately), the explicit requirement that the VMP should be brief, concise and clear was deleted. The range of the VMP was extended to qualification. To the organisational structure already required in the "old" Annex 15 roles and responsibilities for qualification and validation activities were added. This also is new as compared to the draft. The requirement that the ("current" has been deleted as compared to the draft) validation status of the facilities, equipment, systems, processes on site should be summarised was added. Furthermore, the revised Annex 15 requires the description of a deviation management in the validation master plan, the developing of acceptance criteria (in the draft "handling") and the description of a qualification and validation strategy, including re-qualification. As compared to the draft the "ongoing validation strategy" was deleted as well as the requirement to describe a revalidation (substituted by requalification). The assessment of the resources required which was still mentioned in the draft is not required any more. This is also true for the confirmation in the validation master plan that the materials used are of the required quality and that suppliers are qualified to the appropriate level.
New are the last two requirements in this chapter:
- In the context of the quality risk management approach the risk assessments should be repeated in the light of increased knowledge and understanding from any changes. It is explicitly stated that the ways in which risk assessments are used should be clearly documented.
- Checks to ensure the integrity of all data obtained.
Documentation
The second chapter "Documentation" (there is again a reference to the VMP) begins with demanding the use of "good documentation practices" in order to support knowledge management throughout the product lifecycle. All validation documents should be approved and authorised as defined in the quality system. The inter-relationship between documents in complex validation projects should be clearly defined. Validation protocols should be prepared which define the critical systems, attributes and parameters and the associated acceptance criteria. The revised Annex 15 explicitly states the possibility to combine together qualification documents (such as IQ and OQ). Where validation protocols are supplied by a third party the user should confirm their suitability and compliance with internal procedures before approval. The possibility that vendor protocols may be supplemented by additional documentation/test protocols is new as compared to the draft. Any significant changes to the approved protocol (such as acceptance criteria, operating parameters) should be documented as a deviation and be scientifically justified. Results which fail to meet the acceptance criteria should also be recorded as a deviation and be fully investigated according to internal procedures. Any implications for the validation should be discussed in the report. The content of the requirements concerning the report did not change. Cross-references to the protocol are not required any more but the results obtained should be summarised against the acceptance criteria. Review and conclusions should be part of the report.
The final document showed flexibility concerning the formal release for the next stage in the qualification and validation process that can either be part of the validation report approval or as a separate summary document. Completely new is the conditional approval to proceed to the next qualification stage that can be given in the case of deviations for example where there is a documented assessment that there is no significant impact on the next activity.
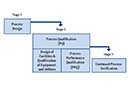
Qualification
The main stages of qualification and some suggested criteria are indicated in the third chapter "Qualification Stages for Equipment, Facilities, Utilities and Systems" as a "could" option. Is this a reference to alternative procedures? The contents of the qualification steps themselves (IQ, OQ, PQ) remained "should" requirements. In the draft they still have been "could" requirements. The definitions for the single qualification steps DQ, IQ and OQ remained unchanged, only the definition for PQ was adjusted. Already at the beginning of the chapter it is indicated that qualification should consider all stages from the development of the user requirements specification (URS) through to the end of use. The specification for new equipment, facilities, utilities or systems should be defined in a user requirements specification and/or a functional specification. Objective is to build in quality at this stage and to mitigate any GMP risks. The URS should be a point of reference throughout the validation life cycle.
Now, DQ is the second step in the qualification. There have been few changes as concerns the content apart from the fact that the requirements of the user requirements specification should be verified during the design qualification.
New parts in the final document are the factory acceptance test (FAT) and the site acceptance test (SAT). A FAT could eventually be carried out for equipment, especially if incorporating novel or complex technology. This is a clear defusing as compared to the draft that still required a FAT as "should". Where appropriate and justified, documentation review and some tests could be performed at the FAT or other stages without the need to repeat on site at IQ/ OQ if it can be shown that the functionality is not affected by the transport and installation. According to the final document a FAT may be supplemented by the execution of a SAT. Prior to installation, equipment should be confirmed to comply with the URS/ functional specification at the vendor site, if applicable.
The requirements concerning the IQ have remained quasi unchanged. Now the verification of the correct installation of components and instrumentation against the engineering drawings and specifications is explicitly required. In the revised Annex 15 verification of the correct installation against pre-defined criteria is now required in an own sub-point.
Depending on the complexity of the equipment, the OQ may now be performed as a combined Installation/Operation Qualification (IOQ). The formal release after the OQ is not required any more. The OQ test should ensure the system is operating as designed. It is not mentioned any more that the calibration should be reviewed at this stage.
For PQ the possibility is stated explicitly that it may (in some cases) be appropriate to perform it in conjunction with OQ or Process Validation. New is the use of worst case batch sizes. Now, the frequency of sampling should be justified. It is new that the extent of PQ tests depends on the results from development.
Re-qualification
Chapter 4 "Re-qualification" is new. The state of qualification should be evaluated at regular intervals and the period should be justified. Furthermore, the possibility of small changes over time should be assessed. The requirement in the draft that where manual processes are used (such as for cleaning), the continued effectiveness of the process should be confirmed at a justified frequency is not contained in this chapter in the final document. Compared to the previously valid Annex 15 the possibility of a review within the re-qualification is not stated any more.
The sub-chapter on the qualification of legacy facilities, systems and equipment was dropped completely.
Chapter 5: Process Validation
It is implicit in this annex that a robust product development process is in place to enable successful process validation. It is explicitly referred to the current EMA Guideline on Process Validation which will also have to be taken into consideration. In so far some parallels can be found between both documents. It is also stated that the GMP requirements go beyond the validation requirements for the documents concerning regulatory submissions. Reference is also made to the lifecycle approach. The traditional approach, a continuous verification approach or a hybrid approach is mentioned as option. Interestingly enough it is indicated that irrespective of the approach used, processes must be shown to be robust and ensure consistent product quality before any product is released to the market. Validation for new products should cover all intended marketed strengths and sites of manufacture. For products which are transferred from one site to another or within the same site, and where there is existing product knowledge a bracketing approach could be used concerning the number of batches, strengths, batch sizes and pack sizes/ container types, if justified. In the case of new products, condition for bracketing is extensive process knowledge from the development stage in conjunction with an appropriate ongoing verification programme.
The identification of critical process parameters and critical quality attributes should be based on a risk assessment. In the case of a site transfer the conditions of the Marketing Authorisation should still be met. Batches manufactured for process validation deviating from the size of commercial scale batches should be justified. The draft still mentions a continuous manufacturing process as example for a deviating batch size. This is not contained in the final document any more. It is explicitly expected that production personnel are involved in the validation activities (product understanding) but personnel from development or site transfer may also be involved. The suppliers of critical starting and packaging materials should be qualified prior to the manufacture of validation batches. Otherwise there should be a written justification based on the application of quality risk management principles. Process knowledge and mathematical models should be available in the case of design spaces. Where validation batches are released to the market, this should be pre-defined. The products should fully comply with GMP and with the marketing authorisation or clinical trial authorisation (reference to Annex 13).
There are very strict rules for concurrent validation which may be carried out only after a justified and strict riskbenefit- assessment. This approach must be documented in the validation master plan and be approved by authorised personnel. There should be sufficient data to support the conclusion that the process is uniform and can meet the defined acceptance criteria. This should be documented and the data should be available to the Qualified Person prior to release of the batch.
When using the traditional approach (manufacture of validation batches under routine conditions), the number of batches and the number of samples taken should be based on quality risk management principles to allow the normal range of variation and trends to be established and to provide sufficient data for evaluation. Each manufacturer must determine and justify the number of batches necessary to demonstrate that the process is capable of consistently delivering quality products.
Without prejudice to the above it is generally considered acceptable that a minimum of three consecutive batches manufactured could constitute a validation of the process, states the text in the revised version. But it is mentioned explicitly that an alternative number of batches may be justified taking into account whether standard methods of manufacture are used and whether similar products or processes are already used. If this way is chosen it should be justified. An initial validation exercise with three batches may need to be supplemented with further data obtained from subsequent batches as part of an on-going process verification exercise. The validation protocol should define the critical process parameters (CPP), critical quality attributes (CQA) and the associated acceptance criteria which should be based on development data or documented process knowledge. The following requirements were added to the validation protocol:
- Summary of the CQAs
- CPPs (with limits)
The protocol should now also contain a summary of noncritical attributes and parameters which will be investigated during the validation activity. This is a difference to the FDA Process Validation Guidance.There should be given reasons for the inclusion of these attributes and parameters in the validation. The method validation of the relevant analyses should be stated as well as the criteria for the process for release, if applicable. There should exist a rationale for the sampling plan. And furthermore, reasons should be given why each in-process control is selected during the validation. But a time table is not required any more.
There are special sub-chapters for "continuous process verification" and "ongoing process verification". The condition for continuous process verification is a quality by design approach, where it has been scientifically established during development that the established control strategy provides a high degree of assurance of product quality. The continuous process verification should naturally be specified in writing and contain a science based control strategy. Process Analytical Technology (PAT) and multivariate statistical process control may be used as tools. Each manufacturer must determine and justify the number of batches necessary to demonstrate that the process is capable of consistently delivering quality products. The same wording is used as for the traditional approach. A hybrid approach using the traditional approach and continuous process verification can also be used. Where there is a substantial amount of product and process knowledge and understanding which has been gained from manufacturing experience and historical batch data, continuous process verification may also be used for any validation activities after changes or during ongoing process verification. This applies even if the product was initially validated using a traditional approach.
In the context of the ongoing process verification product quality should be monitored to be able to demonstrate that a state of control is maintained throughout the product lifecycle with the relevant process trends evaluated. This concerns all possible validation approaches (traditional, continuous or hybrid). The extent and frequency of ongoing process verification should be reviewed periodically and modified if appropriate, according to the level of process understanding. Ongoing process verification should be conducted under an approved protocol or equivalent documents - this is new as compared to the draft - and a corresponding report should be prepared. Statistical tools should be used, where appropriate, to support any conclusions with regard to process variability and capability. The ongoing process verification should also be reflected in the PQR. Incremental changes over time should also be considered and the need for any additional actions, e.g. enhanced sampling, should be assessed. The topic (routine) revalidation has been dropped completely.
Chapter on the verification of transportation
The transport of medicinal products, investigational medicinal products, bulk products and samples should take place in accordance with the conditions defined in the Marketing Authorisation. It is mentioned explicitly, that the verification of transportation (not validation as in the draft) may be challenging due to the variable factors involved. Transportation routes should be clearly defined, however. Seasonal variations should also be considered for transports. The original requirement in the draft that seasonal variations should only be considered for transport across continents has been dropped. A risk assessment should be performed to consider the impact of variables in the transportation process other than those conditions which are continuously controlled or monitored, e.g. delays during transportation, failure of monitoring devices, topping up liquid nitrogen, product susceptibility and any other relevant factors. Continuous monitoring of any critical environmental conditions to which the product may be subjected should be performed.
Chapter on the validation of packaging
Contrary to the draft it is not explicitly stated any more that primary packaging processes should undergo validation. Variation in equipment processing parameters of packaging equipment may have a significant impact on the integrity and correct functioning of the pack, therefore primary and secondary packaging equipment for finished and bulk products should be qualified. Qualification of the equipment used for primary packing should be carried out at the minimum and maximum operating ranges defined for the critical process parameters such as temperature, machine speed and sealing pressure or for any other factors.Chapter on the qualification of utilities
The quality of steam, water, air, other gases etc. should be confirmed following installation using the qualification steps described in the chapter qualification. The requirement in the draft that coolants should also be qualified has been dropped. The period and extent of qualification should also reflect any seasonal variations, if applicable, and the intended use of the utility. A risk assessment should be carried out where there may be direct contact with the product (e.g. HVAC systems) or in order to mitigate the risk of failure due to indirect contact (such as through heat exchangers).
Chapter on the validation of test methods
All analytical test methods used in qualification, validation or cleaning exercises should be validated with an appropriate detection and quantification limit, where necessary, as defined in Chapter 6 of the EU GMP Guidelines Part I. Where microbial testing of product is carried out, the method should be validated to confirm that the product does not influence the recovery of microorganisms. This is also valid for microbial testing of surfaces in clean rooms. They should be validated to confirm that sanitising agents do not influence the recovery of microorganisms.
Chapter on cleaning validation
Chapter 9 "Cleaning Validation" now has more than twice the number of sub-chapters. There were also changes as compared to the draft. Now grouping together of different equipment is mentioned as possible, if the relevant grouping is justified. It is new (also in comparison to the draft) that simulating agents may be used with appropriate scientific justification. The acceptance criteria "visibly clean" as single acceptance criterion is described as not acceptable. Now it is recognised that a cleaning validation programme may take some time to complete and validation with verification after each batch may be required for some products, e.g. investigational medicinal products. There should be sufficient data from the verification to support a conclusion that the equipment is clean. Validation should consider the level of automation in the cleaning process. Automatic processes should be validated as concerns the specified normal operating conditions. Detailed requirements from the draft for manual cleaning, such as the identification of variable factors or worst case approaches have been generalised to all cleaning processes. It is new as compared to the draft that where manual cleaning is performed, the effectiveness of the manual process should be confirmed at a justified frequency. Limits for the carry over of product residues should be based on a toxicological evaluation. The permitted daily exposure (PDE) value should be documented in a risk assessment which includes the relevant references. The removal of any cleaning agents used should also be confirmed. And acceptance criteria should consider the cumulative effect of multiple equipment in the process equipment train. New (also in comparison the draft) is the exemplary reference to (TOC) and conductivity if it is not feasible to test for specific product residues. Dirty and clean-hold times should be defined as part of the cleaning validation. Where campaign manufacture is carried out, the ease of cleaning between batches and the maximum length of a campaign (in both time and number of batches) should be the basis for cleaning validation exercises. The potential for microbial and, or if relevant, endotoxin contamination, should be assessed during validation. The use of worst case products should be based on a scientific rationale and be assessed again if new products are introduced. Criteria for determining the worst case may include solubility, toxicity, (and new compared to the draft) cleanability and potency of the product. Cleaning validation protocols should detail the locations to be sampled and the rationale for the selection. Furthermore, acceptance criteria should be defined. As techniques for sampling, swabbing and/or rinsing or other means depending on the sampling location are stated. The swab material should not influence the result. The following requirement in the draft was dropped: If rinse methods are used, the sampling should be performed during the final rinse in the cleaning procedure. Recovery rates should be defined. Interestingly, the number of validation runs should be based on a risk assessment. The wording in the draft that for investigational medicinal products or products which are only manufactured infrequently, cleaning verification may be used instead of cleaning validation has already been mentioned above in this section. Where a cleaning process is ineffective or is not appropriate for some equipment dedicated equipment should be used for each product (in the draft the reference to chapters 3 and 5 GMP Guideline Part I is missing).
Change Control
The chapter Change Control grew from originally two to seven sub-chapters. Change processes are an important part of knowledge management and should be handled within the pharmaceutical quality system during the life cycle. Changes in the product range, the batch size or the design space now are listed as examples for activities requiring change control. Reference is made to a possible need for any regulatory actions concerning the Marketing Authorisation in the case of a changed design space. Quality risk management should be used to evaluate the potential impact of changes on product quality, pharmaceutical quality systems, documentation, validation, regulatory status, calibration, maintenance and on other systems. This should also help to plan for any necessary process validation, verification or re-qualification efforts. Changes should be authorised and approved by the responsible persons or relevant functional personnel in accordance with the pharmaceutical quality system. Following implementation an evaluation of the effectiveness of change should be carried out. This has already been required after the revision of chapter 1 of the EU GMP Guidelines Part I (valid as of 13 January 2013). More importance than in the draft is attached to supporting data (e.g. copies of documents) to confirm that the impact of the change was demonstrated prior to final approval.Glossary
The following are new definitions in the glossary: bracketing approach, continuous process verification (with reference to ICH Q8), control strategy (with reference to ICH Q10), critical process parameter (with reference to ICH Q8), critical quality attribute (with reference to ICH Q8), design space (with reference to ICH Q8), knowledge management (with reference to ICH Q10), life cycle, ongoing process verification (it is stated explicitly that this term also is known as continued process verification), product realisation (with reference to ICH Q10), quality by design (with reference to ICH Q8), quality risk management (with reference to ICH Q9), state of control, traditional approach, user requirements specification (not included in the draft). In the definition of PQ the connection between facilities, systems and equipment is not explicitly mentioned any more. It is striking that the definition for process validation has remained unchanged in spite of the introduction of the validation lifecycle and that the lifecycle insofar is not part of the definition.
Conclusion:
The revision is very comprehensive. It is questionable how the optional application of the revised Annex 15 can be carried out regarding active substances without introducing additional requirements. The FDA Process Validation Guidance is also mandatory for APIs. The influences of the guidelines ICH Q8, 9 and 10 can be seen clearly, even in the glossary. The alignment with the EMA Guideline on Process Validation, revised in 2014, is also striking. Now the topic design space (ICH Q8) is included in the part on process validation. Now a lot of risk assessments (ICH Q9) are mandatory. And the lifecycle approach and the topic process capabilities (ICH Q10) are included, too. Deviation management has assumed new importance. Third party services are allowed explicitly if the supplier has been qualified accordingly. This can also be seen as an adaptation to reality. It is positive that the preliminary release for the next step (such as qualification) was mentioned for example in the case of deviations if there is a documented assessment that there is no significant impact on the next activity. It was not implemented a clear separation between qualification (with regard to facilities and equipment) and validation (with regard to processes).
New concepts appear without being defined in the glossary, such as FAT and SAT or a functional specification. Retrospective validation and the term (routine) revalidation were dropped completely. But the effectiveness of the manual cleaning processes should be confirmed at a justified frequency. Is this not also a sort of routine revalidation?
Recommendation
14-16 October 2026
Advanced Level: Trending of Process Data for OPV/CPV - Live Online Training
The inclusion of user requirements as separate step and the mention of FAT and SAT - even if merely as could requirement - have made qualification more extensive. FAT and SAT are typical elements of Good Engineering Practice (GEP). Here, the link between GMP and GEP is missing in the document. Can other GEP elements now also be applied (without problems) in the GMP environment? Will even more GEP elements become mandatory in the future?
The greater flexibility as concerns the qualification steps IQ/OQ which now can be carried out together, is a positive element. But this has already been done frequently by industry. There are no in-depth references to alternatives for the qualification, such as ASTM E2500 although the main qualification steps are merely a could option. Unfortunately, information on the qualification of legacy facilities, systems and equipment were dropped completely. It happens again and again that non-GMP environments are becoming GMP environments. May these environments still qualify their old equipment and if so, how?
The reference to transport verification, packaging validation, validation of utilities and validation of analytical methods comes as a surprise. Other regulations are more concrete (such as ICH Q2 (R1) as concerns the validation of analytical methods). The transport verification would probably have better been placed in the context of regulations on Good Distribution Practice. The "old" Annex 15 has been a general guidance to the topic validation/qualification, why has there been this specification? Should not the logical consequence be that validation of the sterilisation process or media fills for example must also be included?
Now, there are three different approaches for process validation, a (modern) continuous verification approach, a traditional approach that is still based on the classical three validation runs and a mixture between both, the hybrid approach. But process robustness has to be established in any case. Despite the accordingly listed restrictions and the necessary justifications as concerns the decision for the number three, the statement that generally at least three consecutive runs are regarded as acceptable is a clear difference to the FDA Guideline on Process Validation. This Guideline doesn't define a number any more. It is positive, that a bracketing approach may be used in justified cases. The possibility of a hybrid approach remains somewhat unclear. This term is not defined in the glossary. But it is mentioned that a change of the validation strategy (from the traditional approach to a verification) is possible with an increasing level of knowledge and understanding of the process. Unfortunately, the possibility of a review in the course of re-qualifications is not explicitly mentioned any more. But could the required evaluation concerning re-qualification not still be carried out as review, where applicable?
As concerns the cleaning validation the criterion "visibly clean" as single acceptance criterion is indicated as not acceptable any more. Now, grouping together of different equipment in the course of the cleaning validation is expressly stated as possibility, if justified. In this case the final version is an adaptation to general practice. Nothing but a toxicological justification (PDE) is targeted as acceptance criterion. This innovation has been expected but it might cause insecurities. The statement that the decision on the number on cleaning validation runs has to be taken on the basis of a risk assessment is very interesting. It was refrained expressly from mentioning the magical number three - in contrast to the process validation. It is good that now a cleaning verification is also possible for products which are only manufactured infrequently. The requirement for dirty and clean hold times constitutes an adaptation to the state-of-the-art.
The chapter Change Control now refers very strongly to regulatory aspects. The requirement for an efficiency control after implementation of a change is new. It has already been required in chapter 1 of the EU GMP Guidelines Part I. Due to the introduction of the feedback loop the term change management would actually have been better for this chapter.
All in all there is an abundance of new requirements, which in part, however, only reproduce the state- of-the-art. Many of the new requirements such as user requirement specifications for the qualification or hold times in the cleaning validation have been used like this by industry for years. The PDE concept is a complete novelty in the cleaning validation. Due to the (necessary) inclusion of ICH Q8-11 and the lifecycle approach the annex has become more holistic, but unfortunately also more ambiguous. Although it is mentioned explicitly, the lifecycle model is not as stringently incorporated in the complete document as in the FDA Guideline on Process Validation. The same is true for the very intensive emphasis on statistics in the FDA Guideline which cannot be found to the same extent in the new Annex 15. Insofar a more strict coordination with the FDA Guideline on Process Validation (also for example as concerns the topic PPQ) would have been desirable.
Author:
Sven Pommeranz
CONCEPT HEIDELBERG

